seqlinkage
Construct phylogenetic tree from pairwise distances
Syntax
PhyloTree = seqlinkage(Distances)
PhyloTree = seqlinkage(Distances, Method)
PhyloTree = seqlinkage(Distances, Method, Names)
Arguments
Distances | Matrix or vector of pairwise distances, such as returned
by the |
Method | Character vector or string that specifies a distance method. Choices are:
|
Names | Specifies alternative labels for leaf nodes. Choices are:
The elements must be unique. The number of elements
must comply with the number of samples used to generate the pairwise
distances in |
Description
PhyloTree = seqlinkage(Distances)Distances,
between the species or products. Distances is
a matrix or vector of pairwise distances, such as returned by the seqpdist function.
PhyloTree = seqlinkage(Distances, Method)
'single' | Nearest distance (single linkage method) |
'complete' | Furthest distance (complete linkage method) |
'average' (default) | Unweighted Pair Group Method Average (UPGMA, group average). |
'weighted' | Weighted Pair Group Method Average (WPGMA) |
'centroid' | Unweighted Pair Group Method Centroid (UPGMC) |
'median' | Weighted Pair Group Method Centroid (WPGMC) |
PhyloTree = seqlinkage(Distances, Method, Names)
Examples
Build Phylogenetic Tree from Pairwise Distances
Create an array of structures representing a multiple alignment of amino acids:
seqs = fastaread('pf00002.fa');Measure the Jukes-Cantor pairwise distances between sequences:
distances = seqpdist(seqs,'method','jukes-cantor','indels','pair');
Build the phylogenetic tree for the multiple sequence alignment from calculated pairwise distances. Specify the method to compute the distances of the new nodes to all other nodes. Provide leaf names:
phylotree = seqlinkage(distances,'single',seqs)Phylogenetic tree object with 32 leaves (31 branches)
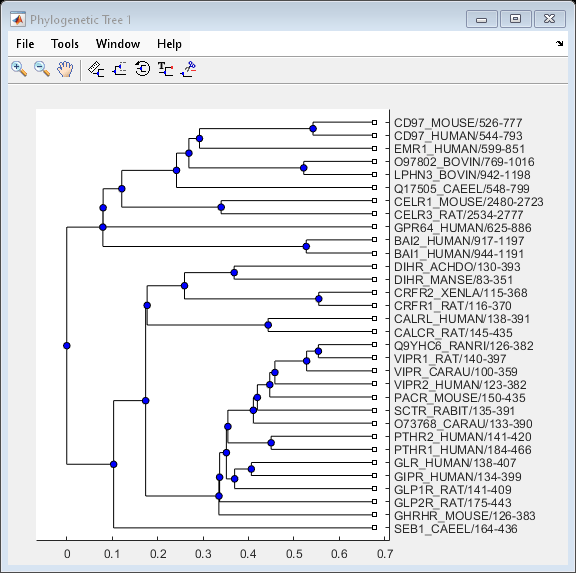
View the phylogenetic tree:
view(phylotree)
Version History
Introduced before R2006aSee Also
phytree | phytreewrite | seqpdist | seqneighjoin | cluster | plot | view
You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)