High-Throughput Sequencing
Gene expression, transcription factor, and methylation analysis of
Next-Generation Sequencing (NGS) data, including RNA-Seq and ChIP-Seq
High-throughput sequencing methods generate large amounts of sequence data and require robust computational tools for further analysis. Bioinformatics Toolbox™ provides algorithms to support common analysis workflows for Next-Generation Sequencing (NGS) data, such as filtering and trimming reads, mapping reads to references, counting the number of reads mapped to genomic features, and performing statistical analyses.
Categories
- Data Import
Import Next-Generation Sequencing (NGS) data and feature annotations from SAM, BAM, FASTA, FASTQ, GTF, and GFF files
- Preprocessing
Manage NGS data with single- and paired-end reads, filter and trim reads, and display quality statistics
- Alignment
Map reads to reference sequences
- Statistical Analysis
Read summarization and statistical analyses on RNA-seq and ChIP-seq data
- Visualization
Visualize alignment of reads to reference sequences
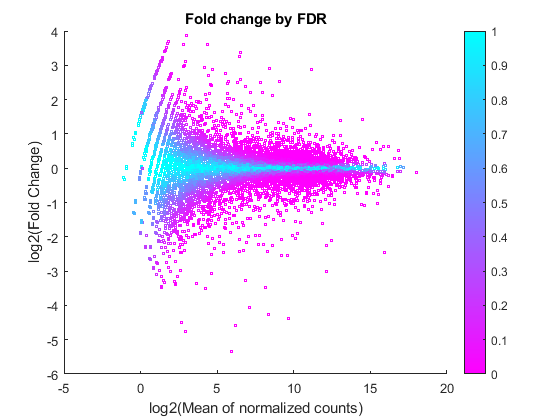
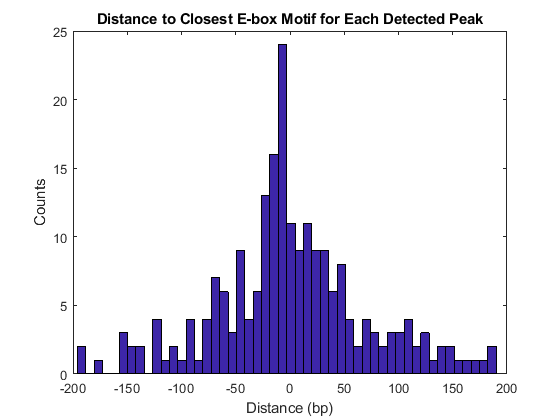
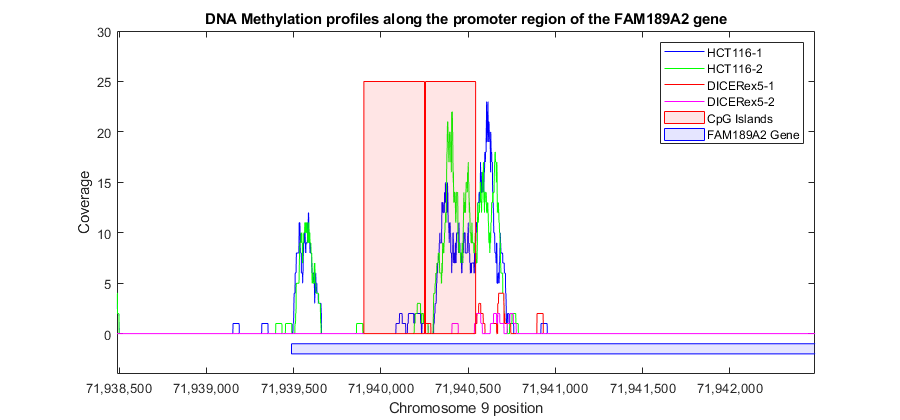
Featured Examples

You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)