Building a Phylogenetic Tree for the Hominidae Species
This example shows how to construct phylogenetic trees from mtDNA sequences for the Hominidae taxa (also known as pongidae). This family embraces the gorillas, chimpanzees, orangutans and humans.
Introduction
The mitochondrial D-loop is one of the fastest mutating sequence regions in animal DNA, and therefore, is often used to compare closely related organisms. The origin of modern man is a highly debated issue that has been addressed by using mtDNA sequences. The limited genetic variability of human mtDNA has been explained in terms of a recent common genetic ancestry, thus implying that all modern-population mtDNAs likely originated from a single woman who lived in Africa less than 200,000 years.
Retrieving Sequence Data from GenBank®
This example uses mitochondrial D-loop sequences isolated for different hominidae species with the following GenBank Accession numbers.
% Species Description GenBank Accession data = {'German_Neanderthal' 'AF011222'; 'Russian_Neanderthal' 'AF254446'; 'European_Human' 'X90314' ; 'Mountain_Gorilla_Rwanda' 'AF089820'; 'Chimp_Troglodytes' 'AF176766'; 'Puti_Orangutan' 'AF451972'; 'Jari_Orangutan' 'AF451964'; 'Western_Lowland_Gorilla' 'AY079510'; 'Eastern_Lowland_Gorilla' 'AF050738'; 'Chimp_Schweinfurthii' 'AF176722'; 'Chimp_Vellerosus' 'AF315498'; 'Chimp_Verus' 'AF176731'; };
You can use the getgenbank function inside a for-loop to retrieve the sequences from the NCBI data repository and load them into MATLAB®.
for ind = 1:length(data) primates(ind).Header = data{ind,1}; primates(ind).Sequence = getgenbank(data{ind,2},'sequenceonly','true'); end
For your convenience, previously downloaded sequences are included in a MAT-file. Note that data in public repositories is frequently curated and updated; therefore, the results of this example might be slightly different when you use up-to-date sequences.
load('primates.mat')
Building a UPGMA Phylogenetic Tree using Distance Methods
Compute pairwise distances using the 'Jukes-Cantor' formula and the phylogenetic tree with the 'UPGMA' distance method. Since the sequences are not pre-aligned, seqpdist performs a pairwise alignment before computing the distances.
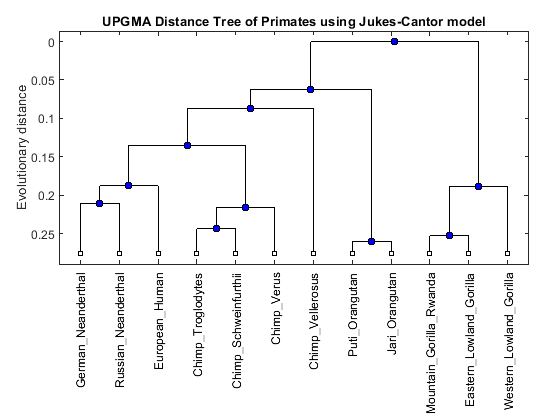
distances = seqpdist(primates,'Method','Jukes-Cantor','Alpha','DNA'); UPGMAtree = seqlinkage(distances,'UPGMA',primates) h = plot(UPGMAtree,'orient','top'); title('UPGMA Distance Tree of Primates using Jukes-Cantor model'); ylabel('Evolutionary distance')
Phylogenetic tree object with 12 leaves (11 branches)
Building a Neighbor-Joining Phylogenetic Tree using Distance Methods
Alternate tree topologies are important to consider when analyzing homologous sequences between species. A neighbor-joining tree can be built using the seqneighjoin function. Neighbor-joining trees use the pairwise distance calculated above to construct the tree. This method performs clustering using the minimum evolution method.
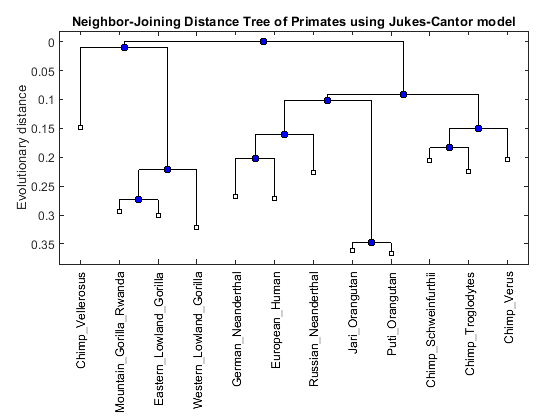
NJtree = seqneighjoin(distances,'equivar',primates) h = plot(NJtree,'orient','top'); title('Neighbor-Joining Distance Tree of Primates using Jukes-Cantor model'); ylabel('Evolutionary distance')
Phylogenetic tree object with 12 leaves (11 branches)
Comparing Tree Topologies
Notice that different phylogenetic reconstruction methods result in different tree topologies. The neighbor-joining tree groups Chimp Vellerosus in a clade with the gorillas, whereas the UPGMA tree groups it near chimps and orangutans. The getcanonical function can be used to compare these isomorphic trees.
sametree = isequal(getcanonical(UPGMAtree), getcanonical(NJtree))
sametree = logical 0
Exploring the UPGMA Phylogenetic Tree
You can explore the phylogenetic tree by considering the nodes (leaves and branches) within a given patristic distance from the 'European Human' entry and reduce the tree to the sub-branches of interest by pruning away non-relevant nodes.
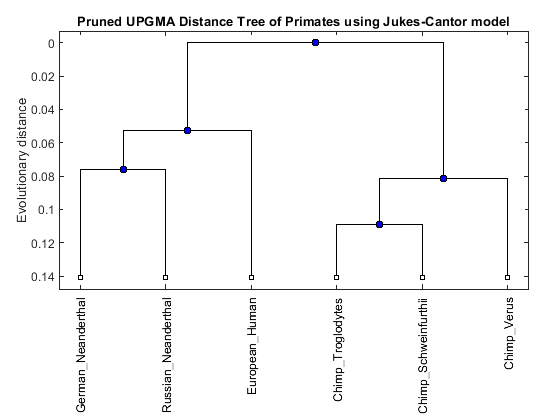
names = get(UPGMAtree,'LeafNames') [h_all,h_leaves] = select(UPGMAtree,'reference',3,'criteria','distance','threshold',0.3); subtree_names = names(h_leaves) leaves_to_prune = ~h_leaves; pruned_tree = prune(UPGMAtree,leaves_to_prune) h = plot(pruned_tree,'orient','top'); title('Pruned UPGMA Distance Tree of Primates using Jukes-Cantor model'); ylabel('Evolutionary distance')
names =
12×1 cell array
{'German_Neanderthal' }
{'Russian_Neanderthal' }
{'European_Human' }
{'Chimp_Troglodytes' }
{'Chimp_Schweinfurthii' }
{'Chimp_Verus' }
{'Chimp_Vellerosus' }
{'Puti_Orangutan' }
{'Jari_Orangutan' }
{'Mountain_Gorilla_Rwanda'}
{'Eastern_Lowland_Gorilla'}
{'Western_Lowland_Gorilla'}
subtree_names =
6×1 cell array
{'German_Neanderthal' }
{'Russian_Neanderthal' }
{'European_Human' }
{'Chimp_Troglodytes' }
{'Chimp_Schweinfurthii'}
{'Chimp_Verus' }
Phylogenetic tree object with 6 leaves (5 branches)
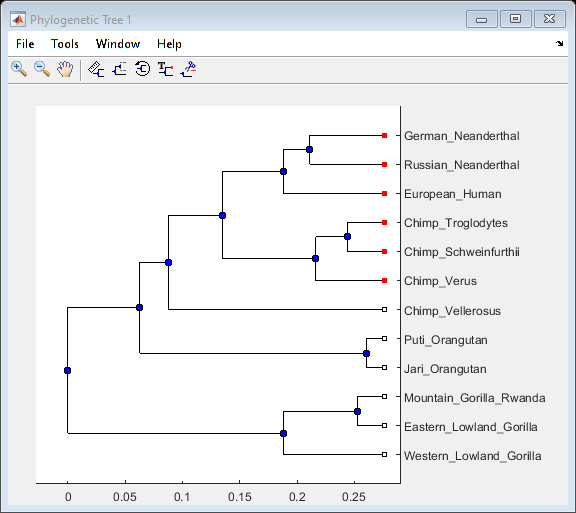
With view you can further explore/edit the phylogenetic tree using an interactive tool. See also phytreeviewer.
view(UPGMAtree,h_leaves)
References
[1] Ovchinnikov, I.V., et al., "Molecular analysis of Neanderthal DNA from the northern Caucasus", Nature, 404(6777):490-3, 2000.
[2] Sajantila, A., et al., "Genes and languages in Europe: an analysis of mitochondrial lineages", Genome Research, 5(1):42-52, 1995.
[3] Krings, M., et al., "Neandertal DNA sequences and the origin of modern humans", Cell, 90(1):19-30, 1997.
[4] Jensen-Seaman, M.I. and Kidd, K.K., "Mitochondrial DNA variation and biogeography of eastern gorillas", Molecular Ecology, 10(9):2241-7, 2001.