rnaseqde
Syntax
Description
diffTable = rnaseqde(countTable,conditionVariables1,conditionVariables2)countTable and performs differential
expression analysis between the conditions (or samples) in
conditionVariables1 and conditionVariables2. The
rows of countTable represent genes (or features), and the columns
represent the conditions. The function first divides each condition by a library size factor
to normalize the count prior to performing the hypothesis test. This size factor is equal to
the median ratio of each feature over the geometric mean of the feature in all conditions.
The function uses an exact test to determine the differences between two groups of counts
that are each assumed to follow a negative binomial distribution [1].
diffTable = rnaseqde(___,Name=Value)
Examples
Use RNA-seq count data that consists of two biological replicates of the control (untreated) samples and two biological replicates of the knock-down (treated) samples [6]. Load the table with read counts for genes.
load("pasilla_count_noMM.mat","geneCountTable")
Display the first few rows of the table.
head(geneCountTable,10)
ID Reference untreated3 untreated4 treated2 treated3
_____________ _________ __________ __________ ________ ________
"FBgn0000003" "3R" 0 1 1 2
"FBgn0000008" "2R" 142 117 138 132
"FBgn0000014" "3R" 20 12 10 19
"FBgn0000015" "3R" 2 4 0 1
"FBgn0000017" "3L" 6591 5127 4809 6027
"FBgn0000018" "2L" 469 530 492 574
"FBgn0000024" "3R" 5 6 10 8
"FBgn0000028" "X" 0 0 2 1
"FBgn0000032" "3R" 1160 1143 1138 1415
"FBgn0000036" "3R" 0 0 0 1
Perform the differential analysis of the control and treated samples using the read count data for genes. Specify both replicates for each condition. The input geneCountTable has an ID column. Optionally, you can append this column to the output table by using IDColumns.
diffTable = rnaseqde(geneCountTable,["untreated3", "untreated4"],... ["treated2", "treated3"],IDColumns="ID"); head(diffTable,5)
ID Mean1 Mean2 Log2FoldChange PValue AdjustedPValue
_____________ _______ _______ ______________ _________ ______________
"FBgn0000003" 0.51415 1.3808 1.4253 1 1
"FBgn0000008" 135.9 129.48 -0.069799 0.67298 0.89231
"FBgn0000014" 16.838 13.384 -0.33119 0.6421 0.87234
"FBgn0000015" 3.1234 0.42413 -2.8806 0.22776 0.57215
"FBgn0000017" 6151.8 5117.4 -0.26559 0.0014429 0.014207
Look at the difference in gene expression between two conditions by displaying the log2 fold change for each gene.
figure scatter(log2(mean([diffTable.Mean1,diffTable.Mean2], 2)),diffTable.Log2FoldChange,3,diffTable.AdjustedPValue,'o') colormap(flipud(cool(256))) colorbar; ylabel("log2(Fold change)") xlabel("log2(Mean of normalized counts)") title("Fold change by FDR")

You can also annotate the values in the plot with the corresponding gene names, interactively select genes, and export gene lists to the workspace.
warnSettings = warning('off','bioinfo:mairplot:ZeroValues'); mairplot(diffTable.Mean2,diffTable.Mean1,Labels=geneCountTable.ID,Type="MA"); set(get(gca,"Xlabel"),"String","mean of normalized counts") set(get(gca,"Ylabel"),"String","log2(fold change)")
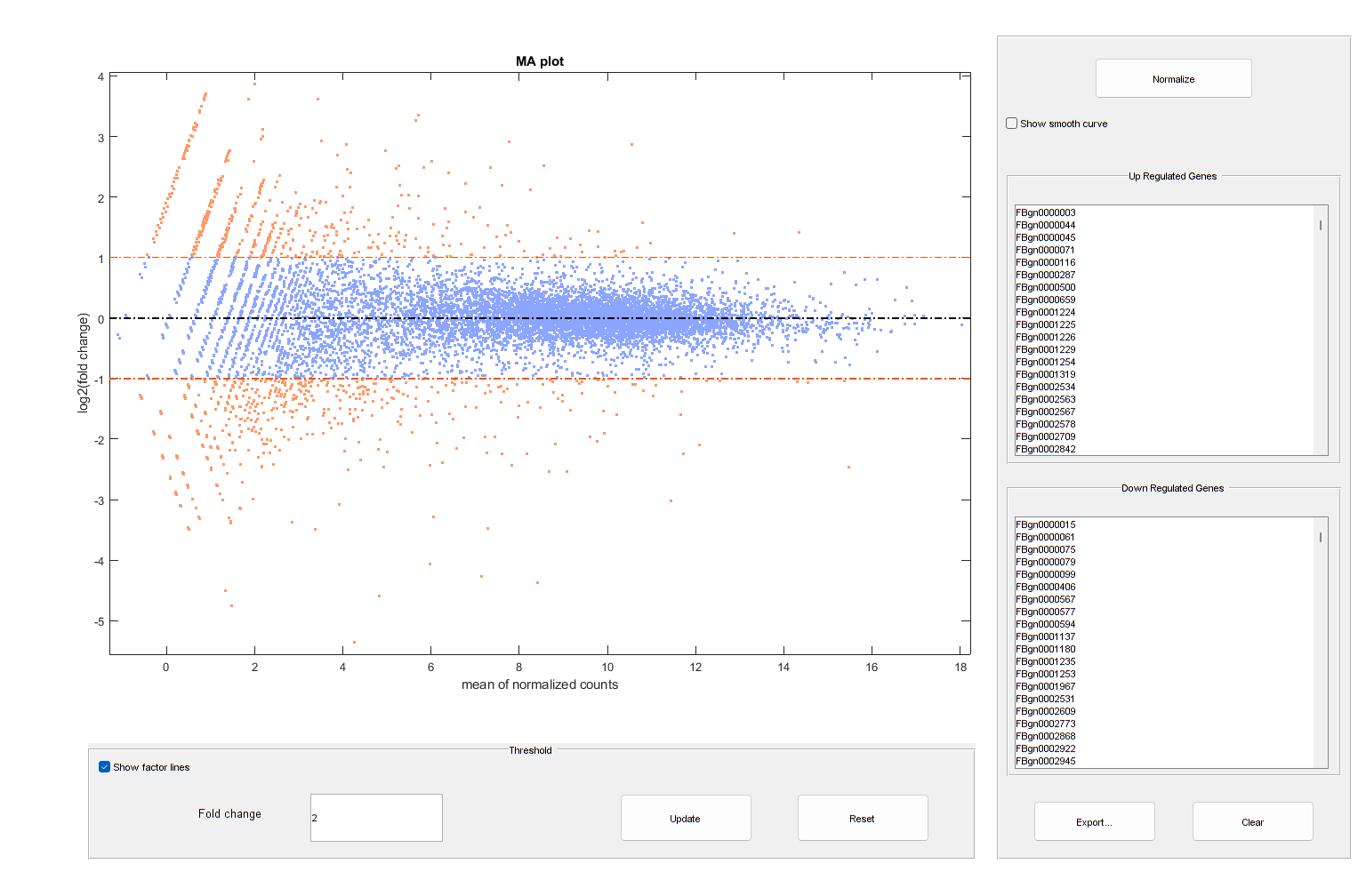
warning(warnSettings);
Input Arguments
Name-Value Arguments
Output Arguments
References
[1] Anders, Simon, and Wolfgang Huber. “Differential Expression Analysis for Sequence Count Data.” Genome Biology 11, no. 10 (October 2010): R106. https://doi.org/10.1186/gb-2010-11-10-r106.
[2] Robinson, Mark D., and Gordon K. Smyth. “Small-Sample Estimation of Negative Binomial Dispersion, with Applications to SAGE Data.” Biostatistics 9, no. 2 (July 11, 2007): 321–32.
[3] Marioni, J. C., C. E. Mason, S. M. Mane, M. Stephens, and Y. Gilad. “RNA-Seq: An Assessment of Technical Reproducibility and Comparison with Gene Expression Arrays.” Genome Research 18, no. 9 (July 30, 2008): 1509–17.
[4] Benjamini, Y., and Hochberg, Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Royal Stat. Soc. 57:289–300.
[5] Storey, John D. “A Direct Approach to False Discovery Rates.” Journal of the Royal Statistical Society: Series B (Statistical Methodology) 64, no. 3 (August 2002): 479–98.
[6] Brooks, A. N., L. Yang, M. O. Duff, K. D. Hansen, J. W. Park, S. Dudoit, S. E. Brenner, and B. R. Graveley. “Conservation of an RNA Regulatory Map between Drosophila and Mammals.” Genome Research 21, no. 2 (February 1, 2011): 193–202.
Version History
Introduced in R2021b