Ground-State Protein Folding Using Variational Quantum Eigensolver (VQE)
This example shows an efficient method for using qubits to encode a protein fold on a 3-D tetrahedral lattice [1], [2]. The ground-state is found through a simulated variational quantum eigensolver (VQE) routine. The VQE algorithm uses classical optimization to improve the initial guess of the ground state, and then a quantum computer calculates the expectation value. The final circuit from the simulation is run on a real QPU for comparison.
The protein is a neuropeptide with seven amino acids, APRLRFY, pictured below. The example assumes a coarse-grained protein model, where "beads" representing amino acids can traverse the lattice and interact with each other. Each bond between amino acids can be in one of four directions, corresponding to corners of a tetrahedron. These four turns can be represented by two qubits.
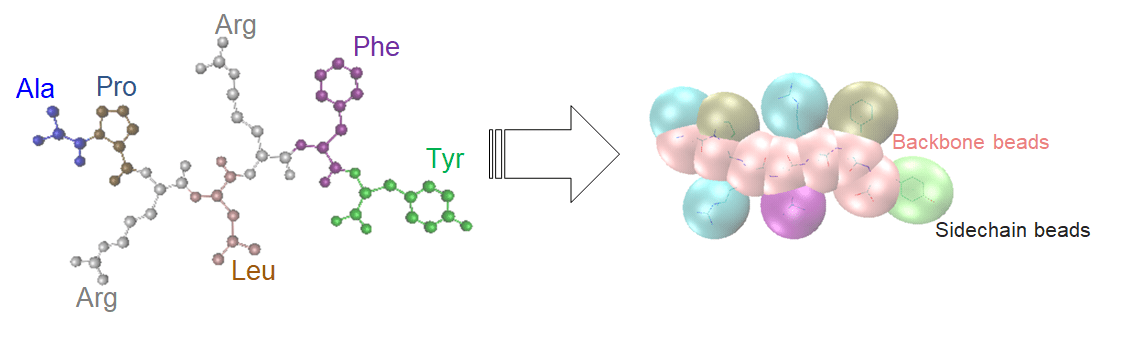
Model Protein Fold with Qubits
The purpose of the embedding methods of [1] and [2] is to model the physical pairwise interaction energy of the protein beads while also imposing energy penalties on unrealistic configurations (such as overlaps or beads on the same lattice site). Therefore, the example is configurable in that you can modify the amino acid letters to test the effects on folding. However, the example code assumes a protein with seven amino acids, so you cannot change the length of the protein string.
Configuration Qubits
A protein of N beads (here N=7) can make N-1 turns, therefore 2*(N-1)=12 bits are required for the APRLRFY protein bonds. However, without loss of generality, the first two turns can be fixed to 01 and 00. One of the bits in the third turn is fixed due to other symmetry considerations. The turn2qubit mapping depicts the 12 bits, denoting the value of the 5 that are fixed, and the 7 that are variable which will be represented by qubits. For details, see the dense encoding scheme in [2].
hyperParams.protein = 'APRLRFY'; hyperParams.turn2qubit = '0100q1qqqqqq'; hyperParams.numQubitsConfig = sum(hyperParams.turn2qubit=='q');
Interaction Qubits
The methods of [1] and [2] can consider interactions between an arbitrary number of nearest-neighbors (NN). This example only considers 1-NN interaction terms, which are only possible between some beads due to the structure of the lattice. Beads separated by less than 5 bonds cannot be 1-NN (Eq. 29 of [2]). There are two interaction pairs in the length-7 protein, between beads 1 and 6, and between beads 2 and 7. Two interaction bits are used to specify whether there is any interaction between the beads in each of these pairs.
hyperParams.numQubitsInteraction = 2;
A matrix of contact energy values is used as a look-up table for pairwise interaction between different amino acids. For this example, the values are chosen at random.
function mat = buildMJInteractions(protein) % Create the MJ interaction energy matrix for the protein, specified with % 1-letter codes N = length(protein); mat = zeros(N,N); rng(29507) MJ = rand(20)*-6; MJ = triu(MJ) + triu(MJ, 1)'; acids = ["C","M","F","I","L","V","W","Y","A","G","T","S","N","Q","D","E","H","R","K","P"]; acid2idx = dictionary(acids, 1:20); for i = 1:N for j=1:N mat(i,j) = MJ(acid2idx(protein(i)), acid2idx(protein(j))); end end end
Call the buildMJInteractions function to find the interaction energies for the protein.
hyperParams.interactionEnergy = buildMJInteractions(hyperParams.protein);
Write Function to Calculate Energy of Folds
The exactHamiltonian function calculates the energy for each possible fold of the protein. Only 1-NN interactions are considered, and energy penalties are imposed for unrealistic configurations.
function energies = exactHamiltonian(bitstrings,hyperParams) % Compute the Hamiltonian for each bit string (i.e., the energy for each fold). % See [2] for details. This does not consider the Hch constraint from % side-chains and the interaction term is only 1-nearest-neighbor (1-NN). % H = Hgc + Hin_1 lambdaDis = 720; % Penalty for interaction distance lambdaLoc = 20; % Penalty for interaction location lambdaBack = 50; % Penalty for unphysical geometry energies = zeros(size(bitstrings,1),1); numBeads = length(hyperParams.protein); for idx = 1:length(energies) bitstring = bitstrings(idx,:); config = hyperParams.turn2qubit; config(config=='q') = bitstring(1:hyperParams.numQubitsConfig); turns = bin2dec(reshape(config,2,numBeads-1)'); %% Geometric Hamiltonian Hgc % Count number of adjacent turns which are equal and impose a penalty for each energies(idx) = lambdaBack*sum(turns(1:end-1) == turns(2:end)); %% 1-NN Interaction Hamiltonian Hin currInteractionQubit = hyperParams.numQubitsConfig; for i=1:(numBeads-4) for j=(i+5):2:numBeads currInteractionQubit = currInteractionQubit+1; if bitstring(currInteractionQubit)=='0' continue; end % Add the interaction energy energies(idx) = energies(idx) + hyperParams.interactionEnergy(i,j); % Compute distances between interacting beads deltaN_ij = zeros(1,4); deltaN_ir = zeros(1,4); deltaN_mj = zeros(1,4); for k=0:3 deltaN_ij(k+1) = (-1).^(i:j-1)*(turns(i:j-1) == k); deltaN_ir(k+1) = (-1).^(i:j-2)*(turns(i:j-2) == k); deltaN_mj(k+1) = (-1).^(i+1:j-1)*(turns(i+1:j-1) == k); end d_ij = norm(deltaN_ij)^2; d_ir = norm(deltaN_ir)^2; d_mj = norm(deltaN_mj)^2; % Add penalty for distance not equal to 1 energies(idx) = energies(idx) + lambdaDis*(d_ij-1); % Add penalty for unphysical nearest neighbour collisons energies(idx) = energies(idx) + lambdaLoc*(2-d_ir); energies(idx) = energies(idx) + lambdaLoc*(2-d_mj); if i-1 >= 1 for k=0:3 deltaN_mj(k+1) = (-1).^(i-1:j-1)*(turns(i-1:j-1) == k); end d_mj = norm(deltaN_mj)^2; energies(idx) = energies(idx) + lambdaLoc*(2-d_mj); end if j+1 <= numBeads for k=0:3 deltaN_ir(k+1) = (-1).^(i:j)*(turns(i:j) == k); end d_ir = norm(deltaN_ir)^2; energies(idx) = energies(idx) + lambdaLoc*(2-d_ir); end end end end end
Compute Minimum Energy for All Folds
Nine qubits are required to fully model a fold (7 configuration qubits and 2 interaction qubits). Use the exactHamiltonian function directly with all possible bit strings (each representing a possible fold), to find the minimum energy fold. You can later compare this value to the result of the quantum-based optimization. This exhaustive search is realistic for small proteins, but is not practical for larger proteins since the number of combinations of folds grows quickly.
hyperParams.numQubitsTotal = hyperParams.numQubitsConfig + hyperParams.numQubitsInteraction; allFolds = dec2bin(0:2^hyperParams.numQubitsTotal-1,hyperParams.numQubitsTotal); allEnergies = exactHamiltonian(allFolds,hyperParams); hyperParams.GroundState.Energy = min(allEnergies); hyperParams.GroundState.Index = find(allEnergies == hyperParams.GroundState.Energy); allFolds(hyperParams.GroundState.Index,:)
ans = 2×9 char array
'101000110'
'101001010'
There are two lowest-energy folds because the last amino acid can occupy either position in the lattice. Find the energy of each of the identified folds.
allEnergies(hyperParams.GroundState.Index)
ans = 2×1
-4.7973
-4.7973
This minimum-energy value matches the interaction energy between beads 1 and 6.
Write CVaR-VQE Objective Function
Next, use a quantum circuit to translate this optimization problem over a set of bit strings into a different optimization problem, this one over a set of angles between -pi and pi. These angles have no connection to the angles between the protein beads. The objective function accepts the set of angles for use inside of the quantum circuit, and the circuit returns various bit strings with different probabilities. Call the exactHamiltonian function on the bit strings returned by the quantum circuit, and use a weighted mean of the smaller energies computed as the value of the objective function.
Specifically, after the energy is computed for each observed fold, the associated probabilities are sorted by energy. The objective function returns an expectation energy computed from the tail end of the probability distribution, cutoff by an alpha parameter. This expectation energy is a conditional value at risk (CVaR). An alpha value of 0.05 was used experimentally in [1], but for a noise-free simulation ProteinVQEObjective uses a smaller cutoff value of 0.025.
function [energy, maxProbFold] = ProteinVQEObjective(parameters,hyperParams) % Construct and simulate the variational circuit ansatz = ProteinConfigAnsatz(parameters); qState = simulate(ansatz); allProbs = (qState.Amplitudes).^2; % There are 10 qubits in the circuit, but only these 9 are used to define a fold foldQubits = [1:8 10]; % Get the most probable fold % Only compute this if second output maxProbFold is requested if nargout > 1 [~,idx] = max(allProbs); maxProbKet = char(qState.BasisStates(idx)); maxProbFold = maxProbKet(foldQubits); end % Sample, and query/get the states and probabilities of the fold qubits qMeasurement = randsample(qState, hyperParams.numShots); [states, probs] = querystates(qMeasurement, foldQubits); % Sort the probabilities by the energy [energies,sort_idx] = sort(exactHamiltonian(char(states), hyperParams)); probs = probs(sort_idx); % Compute CVaR over the low energy tail of the energy distribution, % delimited by a cutoff parameter alpha. alpha = .025; cut_idx = nnz(cumsum(probs) < alpha); cvar_probs = probs(1:cut_idx); cvar_probs(end+1) = alpha - sum(cvar_probs); % Compute expectation energy as the sum of cutoff state energies weighted by their probability energy = dot(cvar_probs, energies(1:cut_idx+1))/alpha; end
Define the number of shots to use in ProteinVQEObjective, and create a function handle that passes in all of the parameter values to ProteinVQEObjective.
hyperParams.numShots = 1024; objFcn = @(theta) ProteinVQEObjective(theta,hyperParams);
Create Circuit Ansatz
Create and plot the variational circuit ansatz for this protein with random angles. Qubits 1-7 represent the configuration, and the other three qubits are for the interaction (including a helper qubit that does not get measured). This circuit can be found as Figure 3 in [1].
function ansatz = ProteinConfigAnsatz(parameters) % Create the circuit ansatz for a 7 amino acid neuropeptide (10 qubit circuit). parameters = reshape(parameters, [2 9]); gates = [hGate([1:7 9 10]) ryGate([1:7 9 10], parameters(1,:)) cxGate(1:4, 2:5) cxGate(5,10) cxGate(10,9) cxGate(9,8) cxGate(8,9) cxGate(9,8) cxGate([8 7 6], [7 6 1]) ryGate([1:8 10], parameters(2,:)) ]; ansatz = quantumCircuit(gates); end
Call ProteinConfigAnsatz with random angles to construct the circuit. Plot the circuit to view the qubits and gates.
ansatz = ProteinConfigAnsatz(rand(2,9)); plot(ansatz)
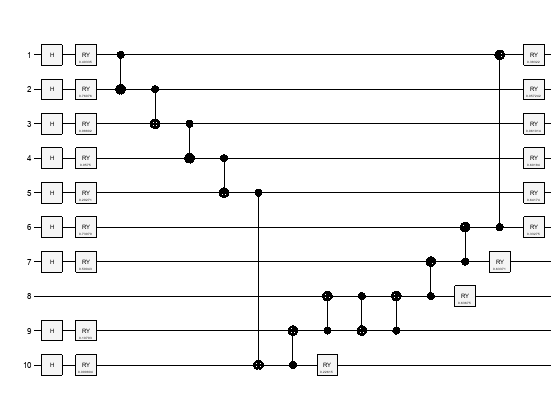
Simulate Iterations of CVaR-VQE
There are two layers of RY rotation gates over the configuration and interaction qubits. These rotation angles are learned parameters in the variational circuit. Use surrogateopt (Global Optimization Toolbox) with the objective function to find the set of RY rotation angles that produce the minimum expectation energy. The original paper [1] uses a genetic algorithm to find the rotation angles. surrogateopt converges to similar values, but because it uses intervals internally, the convergence behavior is comparatively different.
Define the number of angles and set options for the maximum number of function evaluations, plotting function, and initial points. Then call surrogateopt with the objective function, upper and lower bounds for the angles, and options.
numAngles = 2*hyperParams.numQubitsTotal; rng default options = optimoptions("surrogateopt",... "MaxFunctionEvaluations",10, ... "PlotFcn","optimplotfval",... "InitialPoints",pi*ones(numAngles,1)); lb = repmat(-pi,numAngles,1); ub = repmat(pi,numAngles,1); [angles,minEnergy] = surrogateopt(objFcn,lb,ub,[],[],[],[],[],options);
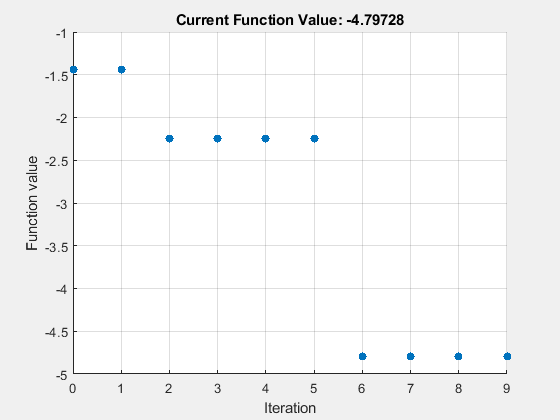
surrogateopt stopped because it exceeded the function evaluation limit set by 'options.MaxFunctionEvaluations'.
Call the objective function using the optimized angles to find the state bit string with highest probability as well as its associated energy.
[groundStateEnergy,groundStateFold] = ProteinVQEObjective(angles,hyperParams)
groundStateEnergy = -4.7973
groundStateFold = '101000110'
This ground-state fold is one of the two folds obtained earlier with the minimum energy calculation.
allFolds(allEnergies==minEnergy,:)
ans = 2×9 char array
'101000110'
'101001010'
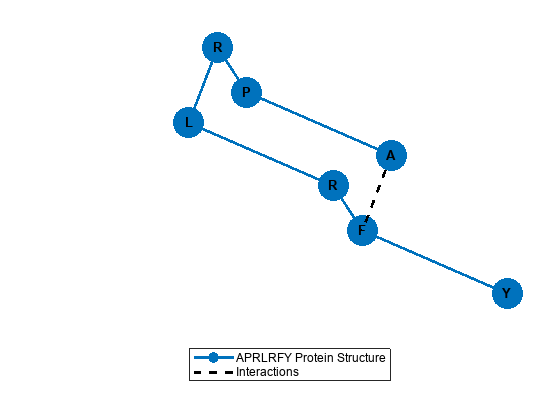
Visualize the ground-state fold using the plotProtein function, which enables you to plot the protein structure from the qubit values.
function plotProtein(bitstring,hyperParams) % Plot protein structure from the bitstring % The input bitstring is expected to be of length 9, with the first 7 bits % specifying turns in direction in the structure, and the last 2 bits % specifying interactions between beads. % Number of beads N = length(hyperParams.protein); % Construct 3D coordinates representing the 4 corners of a tetrahedron % centered in 0. These represent the 4 directions each new bead might % be added on in a tetrahedral grid. turn2bead = ones(4,3); turn2bead(2:4,:) = -1+2*eye(3); % Construct complete bitstring by inserting input bitstring into % the complete string mask. completeBitstring = hyperParams.turn2qubit; completeBitstring(completeBitstring=='q') = bitstring(1:hyperParams.numQubitsConfig); % Each pair of bits is converted into a number between 0 and 3 turns = bin2dec(reshape(completeBitstring,2,[])'); % Change direction in which we follow the tetrahedral grid in each bead % on the line. signs = (-1).^(0:N-1)'; % Compute placements of each bead. beads = cumsum(signs.*[zeros(1,3);turn2bead(turns+1,:)]); % Plot the beads connecting by lines, and add a text label to each figure plot3(beads(:,1),beads(:,2),beads(:,3),'.-','LineWidth',2,'MarkerSize',80,'SeriesIndex',1) axis off viewDir = -[6 4 1]; view(viewDir); % Plot text, ensuring it is in front of the lines beadsText = beads + 0.03*viewDir; text(beadsText(:,1),beadsText(:,2),beadsText(:,3),hyperParams.protein', ... 'FontWeight', 'bold', 'HorizontalAlignment','center') % Whether there are interactions between any pair of beads is determined % by the additional bits in the string. interactions = []; currInteractionQubit = hyperParams.numQubitsConfig+1; for i=1:(N-5) for j=(i+5):2:N if bitstring(currInteractionQubit) == '1' interactions = [interactions;beads(i,:);beads(j,:);nan*ones(1,3)]; %#ok<AGROW> end currInteractionQubit = currInteractionQubit+1; end end if ~isempty(interactions) hold on plot3(interactions(:,1),interactions(:,2),interactions(:,3),'k--','LineWidth',2) hold off legend([hyperParams.protein+" Protein Structure";"Interactions"], "Location","southoutside") else legend(hyperParams.protein+" Protein Structure", "Location","southoutside") end end
The protein has two possible interactions considered in the 1-NN model. The fold with the lowest energy only has one of these interactions. Use plotProtein to visualize the lowest energy fold.
plotProtein(groundStateFold,hyperParams)
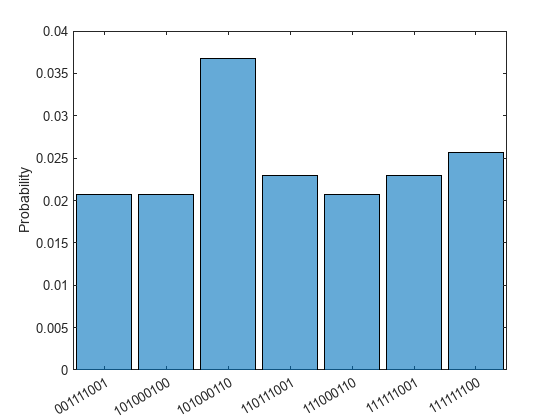
Next, construct the quantum circuit using the optimized angles and simulate the circuit to see the expected probability distribution over states. Use a threshold to filter out states with probabilities less than 2%. The ground-state fold 101000110 appears again as the state with the highest probability.
optimized_circuit = ProteinConfigAnsatz(angles); sv = simulate(optimized_circuit); histogram(sv,[1:8 10],Threshold=0.02)
Run Final Iteration on QPU
Connect to the IonQ device using quantum.backend.QuantumDeviceAWS. Specify the region of the device and path to a bucket to store results.
reg = "us-east-1"; bucketPath = "s3://amazon-braket-mathworks/doc-examples"; device = quantum.backend.QuantumDeviceAWS("Harmony",S3Path=bucketPath,Region=reg)
device =
QuantumDeviceAWS with properties:
Name: "Harmony"
DeviceARN: "arn:aws:braket:us-east-1::device/qpu/ionq/Harmony"
Region: "us-east-1"
S3Path: "s3://amazon-braket-mathworks/doc-examples"
Create a task to run the circuit ansatz with optimized angles on the QPU. Specify the number of shots as 500.
task = run(optimized_circuit,device,NumShots=500); wait(task)
Fetch the results and plot a histogram of the states for all but the ninth qubit. Specify a lower threshold of 2% to filter out unlikely states.
results = fetchOutput(task); histogram(results,[1:8 10],Threshold=0.02)
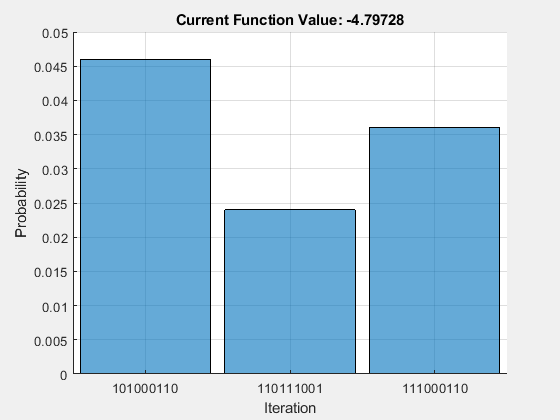
The ground-state fold 101000110 appears again as the state with the highest probability, so the QPU results agree with the local simulation of the circuit.
References
[1] Robert, Anton, Panagiotis Kl. Barkoutsos, Stefan Woerner, and Ivano Tavernelli. “Resource-Efficient Quantum Algorithm for Protein Folding.” Npj Quantum Information 7, no. 1 (February 17, 2021): 38. https://doi.org/10.1038/s41534-021-00368-4.
[2] Robert, Anton, Panagiotis Kl. Barkoutsos, Stefan Woerner, and Ivano Tavernelli. “Supplementary Information for 'Resource-Efficient Quantum Algorithm for Protein Folding'" Npj Quantum Information 7, no. 1 (February 17, 2021): 38. https://doi.org/10.1038/s41534-021-00368-4.
See Also
Topics
- Introduction to Quantum Computing
- Run Quantum Circuit on Hardware Using AWS
- Graph Coloring with Grover's Algorithm
- Solve XOR Problem Using Quantum Neural Network (QNN)
- Quantum Monte Carlo (QMC) Simulation