isotopicdist
Calculate high-resolution isotope mass distribution and density function
Syntax
Description
[ analyzes a compound specified by an
empirical chemical formula represented by the structure MD,Info,DF]
= isotopicdist(Formula)Formula. The
field names in Formula must be valid element symbols and are case
sensitive. The respective values in Formula are the number of atoms for
each element. Formula can also be an array of structures that specifies
multiple formulas. The field names can be in any order within a structure. However, if there
are multiple structures, the order must be the same in each.
[ calculate
high-resolution isotope mass distribution and density function using one or more
MD,Info,DF]
= isotopicdist(___,Name,Value)Name,Value arguments. Use name-value arguments with any combination
of arguments from the previous syntaxes.
Examples
Calculate and display the isotopic mass distribution of the peptide sequence MATLAP with an Acetyl N-terminal and an Amide C-terminal:ю
MD = isotopicdist("MATLAP", ... NTerminal="acetyl", ... CTerminal="amide", ... ShowPlot=true)
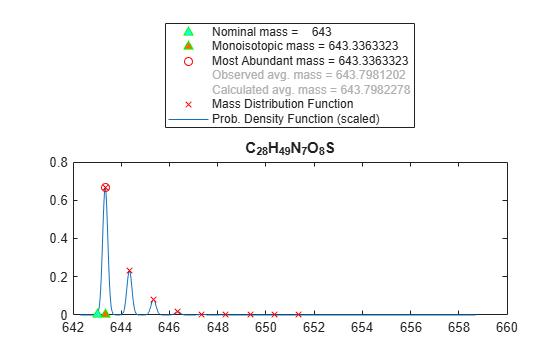
MD = 9×2
643.3363 0.6676
644.3388 0.2306
645.3378 0.0797
646.3386 0.0181
647.3396 0.0033
648.3409 0.0005
649.3423 0.0001
650.3439 0.0000
651.3455 0.0000
Calculate and display the isotopic mass distribution of Glutamine ():
MD = isotopicdist([5 10 2 3 0],ShowPlot=true)

MD = 5×2
146.0691 0.9328
147.0715 0.0595
148.0733 0.0074
149.0755 0.0004
150.0774 0.0000
Display the isotopic mass distribution of the "averagine" model, whose molecular formula represents the statistical occurrences of amino acids from all known proteins.
isotopicdist([4.9384 7.7583 1.3577 1.4773 0.0417])

Input Arguments
Name-Value Arguments
Output Arguments
More About
References
[1] Rockwood, A. L., S. L. Van Orden, and R. D. Smith. "Rapid Calculation of Isotope Distributions." Anal. Chem. 67:15 (1995): 2699–2704.
[2] Rockwood, A. L., S. L. Van Orden, and R. D. Smith. "Ultrahigh Resolution Isotope Distribution Calculations." Rapid Commun. Mass Spectrum 10 (1996): 54–59.
[3] Senko, M.W., S. C. Beu, and F. W. McLafferty. "Automated assignment of charge states from resolved isotopic peaks for multiply charged ions." J. Am. Soc. Mass Spectrom. 6 (1995): 52–56.
[4] Senko, M.W., S. C. Beu, and F. W. McLafferty. "Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions." J. Am. Soc. Mass Spectrom. 6 (1995): 229–233.
Version History
Introduced in R2009b
See Also
cleave | getgenpept | genpeptread | int2aa | nt2aa | aminolookup | cleavelookup | molweight