TreeBagger
Ensemble of bagged decision trees
Description
A TreeBagger object is an ensemble of bagged decision trees for
either classification or regression. Individual decision trees tend to overfit.
Bagging, which stands for bootstrap aggregation, is an ensemble method that
reduces the effects of overfitting and improves generalization.
Creation
The TreeBagger function grows every tree in the
TreeBagger ensemble model using bootstrap samples of the input data.
Observations not included in a sample are considered "out-of-bag" for that tree. The function
selects a random subset of predictors for each decision split by using the random forest
algorithm [1].
Syntax
Description
Tip
By default, the TreeBagger function grows classification decision
trees. To grow regression decision trees, specify the name-value argument
Method as "regression".
Mdl = TreeBagger(NumTrees,Tbl,ResponseVarName)Mdl) of NumTrees bagged
classification trees, trained by the predictors in the table Tbl and the
class labels in the variable Tbl.ResponseVarName.
Mdl = TreeBagger(NumTrees,Tbl,formula)Mdl trained by the predictors in the table
Tbl. The input formula is an explanatory model of
the response and a subset of predictor variables in Tbl used to fit
Mdl. Specify formula using Wilkinson Notation.
Mdl = TreeBagger(___,Name=Value)Mdl with additional options specified by one or more name-value
arguments, using any of the previous input argument combinations. For example, you can specify
the algorithm used to find the best split on a categorical predictor by using the name-value
argument PredictorSelection.
Input Arguments
Name-Value Arguments
Output Arguments
Properties
Object Functions
Examples
Create an ensemble of bagged classification trees for Fisher's iris data set. Then, view the first grown tree, plot the out-of-bag classification error, and predict labels for out-of-bag observations.
Load the fisheriris data set. Create X as a numeric matrix that contains four measurements for 150 irises. Create Y as a cell array of character vectors that contains the corresponding iris species.
load fisheriris
X = meas;
Y = species;Set the random number generator to default for reproducibility.
rng("default")Train an ensemble of bagged classification trees using the entire data set. Specify 50 weak learners. Store the out-of-bag observations for each tree. By default, TreeBagger grows deep trees.
Mdl = TreeBagger(50,X,Y,... Method="classification",... OOBPrediction="on")
Mdl =
TreeBagger
Ensemble with 50 bagged decision trees:
Training X: [150x4]
Training Y: [150x1]
Method: classification
NumPredictors: 4
NumPredictorsToSample: 2
MinLeafSize: 1
InBagFraction: 1
SampleWithReplacement: 1
ComputeOOBPrediction: 1
ComputeOOBPredictorImportance: 0
Proximity: []
ClassNames: 'setosa' 'versicolor' 'virginica'
Properties, Methods
Mdl is a TreeBagger ensemble for classification trees.
The Mdl.Trees property is a 50-by-1 cell vector that contains the trained classification trees for the ensemble. Each tree is a CompactClassificationTree object. View the graphical display of the first trained classification tree.
view(Mdl.Trees{1},Mode="graph")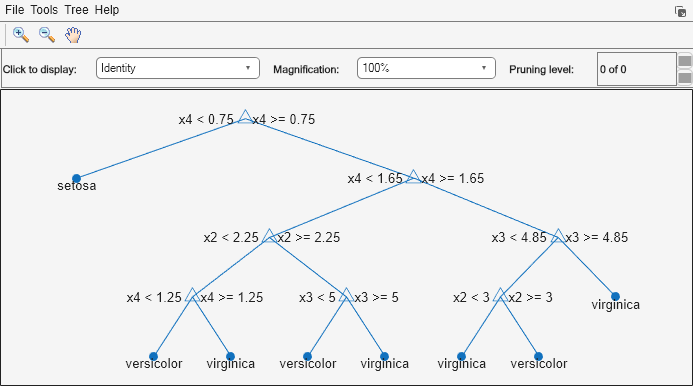
Plot the out-of-bag classification error over the number of grown classification trees.
plot(oobError(Mdl)) xlabel("Number of Grown Trees") ylabel("Out-of-Bag Classification Error")
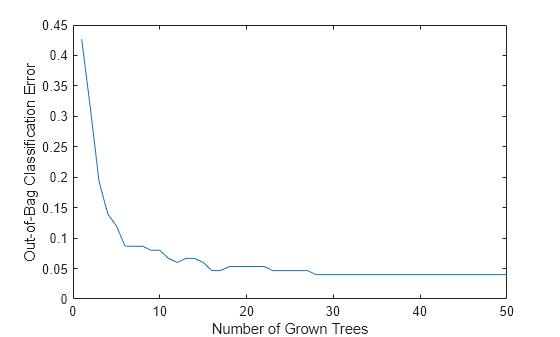
The out-of-bag error decreases as the number of grown trees increases.
Predict labels for out-of-bag observations. Display the results for a random set of 10 observations.
oobLabels = oobPredict(Mdl); ind = randsample(length(oobLabels),10); table(Y(ind),oobLabels(ind),... VariableNames=["TrueLabel" "PredictedLabel"])
ans=10×2 table
TrueLabel PredictedLabel
______________ ______________
{'setosa' } {'setosa' }
{'virginica' } {'virginica' }
{'setosa' } {'setosa' }
{'virginica' } {'virginica' }
{'setosa' } {'setosa' }
{'virginica' } {'virginica' }
{'setosa' } {'setosa' }
{'versicolor'} {'versicolor'}
{'versicolor'} {'virginica' }
{'virginica' } {'virginica' }
Create an ensemble of bagged regression trees for the carsmall data set. Then, predict conditional mean responses and conditional quartiles.
Load the carsmall data set. Create X as a numeric vector that contains the car engine displacement values. Create Y as a numeric vector that contains the corresponding miles per gallon.
load carsmall
X = Displacement;
Y = MPG;Set the random number generator to default for reproducibility.
rng("default")Train an ensemble of bagged regression trees using the entire data set. Specify 100 weak learners.
Mdl = TreeBagger(100,X,Y,... Method="regression")
Mdl =
TreeBagger
Ensemble with 100 bagged decision trees:
Training X: [94x1]
Training Y: [94x1]
Method: regression
NumPredictors: 1
NumPredictorsToSample: 1
MinLeafSize: 5
InBagFraction: 1
SampleWithReplacement: 1
ComputeOOBPrediction: 0
ComputeOOBPredictorImportance: 0
Proximity: []
Properties, Methods
Mdl is a TreeBagger ensemble for regression trees.
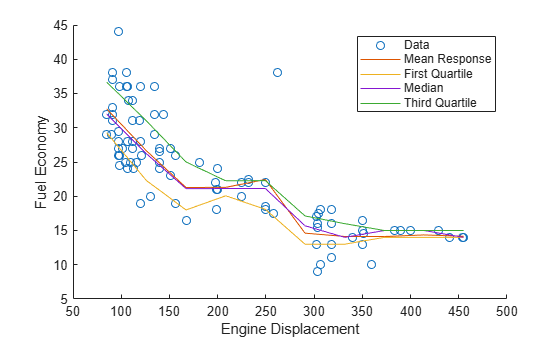
For 10 equally spaced engine displacements between the minimum and maximum in-sample displacement, predict conditional mean responses (YMean) and conditional quartiles (YQuartiles).
predX = linspace(min(X),max(X),10)';
YMean = predict(Mdl,predX);
YQuartiles = quantilePredict(Mdl,predX,...
Quantile=[0.25,0.5,0.75]);Plot the observations, estimated mean responses, and estimated quartiles.
hold on plot(X,Y,"o"); plot(predX,YMean) plot(predX,YQuartiles) hold off ylabel("Fuel Economy") xlabel("Engine Displacement") legend("Data","Mean Response",... "First Quartile","Median",..., "Third Quartile")
Create two ensembles of bagged regression trees, one using the standard CART algorithm for splitting predictors, and the other using the curvature test for splitting predictors. Then, compare the predictor importance estimates for the two ensembles.
Load the carsmall data set and convert the variables Cylinders, Mfg, and Model_Year to categorical variables. Then, display the number of categories represented in the categorical variables.
load carsmall
Cylinders = categorical(Cylinders);
Mfg = categorical(cellstr(Mfg));
Model_Year = categorical(Model_Year);
numel(categories(Cylinders))ans = 3
numel(categories(Mfg))
ans = 28
numel(categories(Model_Year))
ans = 3
Create a table that contains eight car metrics.
Tbl = table(Acceleration,Cylinders,Displacement,...
Horsepower,Mfg,Model_Year,Weight,MPG);Set the random number generator to default for reproducibility.
rng("default")Train an ensemble of 200 bagged regression trees using the entire data set. Because the data has missing values, specify to use surrogate splits. Store the out-of-bag information for predictor importance estimation.
By default, TreeBagger uses the standard CART, an algorithm for splitting predictors. Because the variables Cylinders and Model_Year each contain only three categories, the standard CART prefers splitting a continuous predictor over these two variables.
MdlCART = TreeBagger(200,Tbl,"MPG",... Method="regression",Surrogate="on",... OOBPredictorImportance="on");
TreeBagger stores predictor importance estimates in the property OOBPermutedPredictorDeltaError.
impCART = MdlCART.OOBPermutedPredictorDeltaError;
Train a random forest of 200 regression trees using the entire data set. To grow unbiased trees, specify to use the curvature test for splitting predictors.
MdlUnbiased = TreeBagger(200,Tbl,"MPG",... Method="regression",Surrogate="on",... PredictorSelection="curvature",... OOBPredictorImportance="on"); impUnbiased = MdlUnbiased.OOBPermutedPredictorDeltaError;
Create bar graphs to compare the predictor importance estimates impCART and impUnbiased for the two ensembles.
tiledlayout(1,2,Padding="compact"); nexttile bar(impCART) title("Standard CART") ylabel("Predictor Importance Estimates") xlabel("Predictors") h = gca; h.XTickLabel = MdlCART.PredictorNames; h.XTickLabelRotation = 45; h.TickLabelInterpreter = "none"; nexttile bar(impUnbiased); title("Curvature Test") ylabel("Predictor Importance Estimates") xlabel("Predictors") h = gca; h.XTickLabel = MdlUnbiased.PredictorNames; h.XTickLabelRotation = 45; h.TickLabelInterpreter = "none";
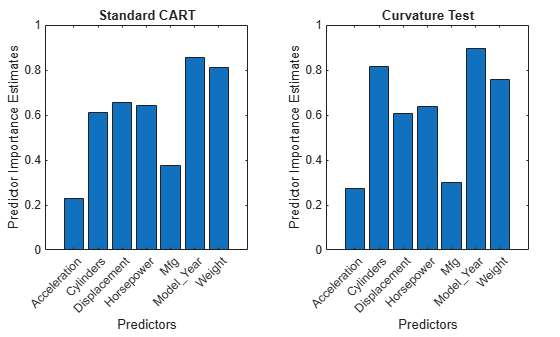
For the CART model, the continuous predictor Weight is the second most important predictor. For the unbiased model, the predictor importance of Weight is smaller in value and ranking.
Train an ensemble of bagged classification trees for observations in a tall array, and find the misclassification probability of each tree in the model for weighted observations. This example uses the data set airlinesmall.csv, a large data set that contains a tabular file of airline flight data.
When you perform calculations on tall arrays, MATLAB® uses either a parallel pool (default if you have Parallel Computing Toolbox™) or the local MATLAB session. To run the example using the local MATLAB session when you have Parallel Computing Toolbox, change the global execution environment by using the mapreducer function.
mapreducer(0)
Create a datastore that references the location of the folder containing the data set. Select a subset of the variables to work with, and treat "NA" values as missing data so that the datastore function replaces them with NaN values. Create the tall table tt to contain the data in the datastore.
ds = datastore("airlinesmall.csv"); ds.SelectedVariableNames = ["Month" "DayofMonth" "DayOfWeek",... "DepTime" "ArrDelay" "Distance" "DepDelay"]; ds.TreatAsMissing = "NA"; tt = tall(ds)
tt =
M×7 tall table
Month DayofMonth DayOfWeek DepTime ArrDelay Distance DepDelay
_____ __________ _________ _______ ________ ________ ________
10 21 3 642 8 308 12
10 26 1 1021 8 296 1
10 23 5 2055 21 480 20
10 23 5 1332 13 296 12
10 22 4 629 4 373 -1
10 28 3 1446 59 308 63
10 8 4 928 3 447 -2
10 10 6 859 11 954 -1
: : : : : : :
: : : : : : :
Determine the flights that are late by 10 minutes or more by defining a logical variable that is true for a late flight. This variable contains the class labels Y. A preview of this variable includes the first few rows.
Y = tt.DepDelay > 10
Y = M×1 tall logical array 1 0 1 1 0 1 0 0 : :
Create a tall array X for the predictor data.
X = tt{:,1:end-1}X =
M×6 tall double matrix
10 21 3 642 8 308
10 26 1 1021 8 296
10 23 5 2055 21 480
10 23 5 1332 13 296
10 22 4 629 4 373
10 28 3 1446 59 308
10 8 4 928 3 447
10 10 6 859 11 954
: : : : : :
: : : : : :
Create a tall array W for the observation weights by arbitrarily assigning double weights to the observations in class 1.
W = Y+1;
Remove the rows in X, Y, and W that contain missing data.
R = rmmissing([X Y W]); X = R(:,1:end-2); Y = R(:,end-1); W = R(:,end);
Train an ensemble of 20 bagged classification trees using the entire data set. Specify a weight vector and uniform prior probabilities. For reproducibility, set the seeds of the random number generators using rng and tallrng. The results can vary depending on the number of workers and the execution environment for the tall arrays. For details, see Control Where Your Code Runs.
rng("default") tallrng("default") tMdl = TreeBagger(20,X,Y,... Weights=W,Prior="uniform")
Evaluating tall expression using the Local MATLAB Session: - Pass 1 of 1: Completed in 0.44 sec Evaluation completed in 0.47 sec Evaluating tall expression using the Local MATLAB Session: - Pass 1 of 1: Completed in 1.5 sec Evaluation completed in 1.6 sec Evaluating tall expression using the Local MATLAB Session: - Pass 1 of 1: Completed in 3.8 sec Evaluation completed in 3.8 sec
tMdl =
CompactTreeBagger
Ensemble with 20 bagged decision trees:
Method: classification
NumPredictors: 6
ClassNames: '0' '1'
Properties, Methods
tMdl is a CompactTreeBagger ensemble with 20 bagged decision trees. For tall data, the TreeBagger function returns a CompactTreeBagger object.
Calculate the misclassification probability of each tree in the model. Attribute a weight contained in the vector W to each observation by using the Weights name-value argument.
terr = error(tMdl,X,Y,Weights=W)
Evaluating tall expression using the Local MATLAB Session: - Pass 1 of 1: Completed in 4.7 sec Evaluation completed in 4.7 sec
terr = 20×1
0.1420
0.1214
0.1115
0.1078
0.1037
0.1027
0.1005
0.0997
0.0981
0.0983
⋮
Find the average misclassification probability for the ensemble of decision trees.
avg_terr = mean(terr)
avg_terr = 0.1022
More About
Tips
For a
TreeBaggermodelMdl, theTreesproperty contains a cell vector ofMdl.NumTreesCompactClassificationTreeorCompactRegressionTreeobjects. View the graphical display of thetgrown tree by entering:view(Mdl.Trees{t})For regression problems,
TreeBaggersupports mean and quantile regression (that is, quantile regression forest [5]).To predict mean responses or estimate the mean squared error given data, pass a
TreeBaggermodel object and the data topredictorerror, respectively. To perform similar operations for out-of-bag observations, useoobPredictoroobError.To estimate quantiles of the response distribution or the quantile error given data, pass a
TreeBaggermodel object and the data toquantilePredictorquantileError, respectively. To perform similar operations for out-of-bag observations, useoobQuantilePredictoroobQuantileError.
Standard CART tends to select split predictors containing many distinct values, such as continuous variables, over those containing few distinct values, such as categorical variables [4]. Consider specifying the curvature or interaction test if either of the following is true:
The data has predictors with relatively fewer distinct values than other predictors; for example, the predictor data set is heterogeneous.
Your goal is to analyze predictor importance.
TreeBaggerstores predictor importance estimates in theOOBPermutedPredictorDeltaErrorproperty.
For more information on predictor selection, see the name-value argument
PredictorSelectionfor classification trees or the name-value argumentPredictorSelectionfor regression trees.
Algorithms
If you specify the
Cost,Prior, andWeightsname-value arguments, the output model object stores the specified values in theCost,Prior, andWproperties, respectively. TheCostproperty stores the user-specified cost matrix (C) without modification. ThePriorandWproperties store the prior probabilities and observation weights, respectively, after normalization. For model training, the software updates the prior probabilities and observation weights to incorporate the penalties described in the cost matrix. For details, see Misclassification Cost Matrix, Prior Probabilities, and Observation Weights.The
TreeBaggerfunction generates in-bag samples by oversampling classes with large misclassification costs and undersampling classes with small misclassification costs. Consequently, out-of-bag samples have fewer observations from classes with large misclassification costs and more observations from classes with small misclassification costs. If you train a classification ensemble using a small data set and a highly skewed cost matrix, then the number of out-of-bag observations per class might be very low. Therefore, the estimated out-of-bag error might have a large variance and be difficult to interpret. The same phenomenon can occur for classes with large prior probabilities.For details on how the
TreeBaggerfunction selects split predictors, and for information on node-splitting algorithms when the function grows decision trees, see Algorithms for classification trees and Algorithms for regression trees.
Alternative Functionality
Statistics and Machine Learning Toolbox™ offers three objects for bagging and random forest:
ClassificationBaggedEnsembleobject created by thefitcensemblefunction for classificationRegressionBaggedEnsembleobject created by thefitrensemblefunction for regressionTreeBaggerobject created by theTreeBaggerfunction for classification and regression
For details about the differences between TreeBagger and
bagged ensembles (ClassificationBaggedEnsemble and
RegressionBaggedEnsemble), see Comparison of TreeBagger and Bagged Ensembles.
References
[1] Breiman, Leo. "Random Forests." Machine Learning 45 (2001): 5–32. https://doi.org/10.1023/A:1010933404324.
[2] Breiman, Leo, Jerome Friedman, Charles J. Stone, and R. A. Olshen. Classification and Regression Trees. Boca Raton, FL: CRC Press, 1984.
[3] Loh, Wei-Yin. "Regression Trees with Unbiased Variable Selection and Interaction Detection." Statistica Sinica 12, no. 2 (2002): 361–386. https://www.jstor.org/stable/24306967.
[4] Loh, Wei-Yin, and Yu-Shan Shih. "Split Selection for Classification Trees." Statistica Sinica 7, no. 4 (1997): 815–840. https://www.jstor.org/stable/24306157.
[5] Meinshausen, Nicolai. "Quantile Regression Forests." Journal of Machine Learning Research 7, no. 35 (2006): 983–999. https://jmlr.org/papers/v7/meinshausen06a.html.
[6] Genuer, Robin, Jean-Michel Poggi, Christine Tuleau-Malot, and Nathalie Villa-Vialanei. "Random Forests for Big Data." Big Data Research 9 (2017): 28–46. https://doi.org/10.1016/j.bdr.2017.07.003.